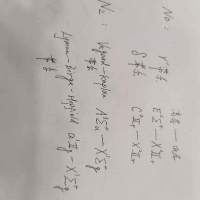小木虫
登陆
|
注册
首页
导读
期刊
发现
社区
招聘老师
当前位置:
小木虫首页
>> 量子化学
Chem3D运行Gamess预测红外图谱没反应咋回事呀
颜朽儿
按照网上的教程做的,但是运行了一个多小时没有一点进展(不是卡顿,正在运行中)------------GAMESSInterface------------Model:Untitled-1GAMESSJob:PredictIR/RamanSpectrumRHF/...
量子化学
150
3
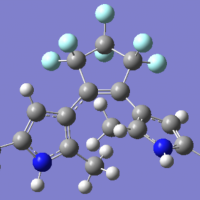
求助:金属团簇Fe-NU-1000所带电荷及自旋多重度的判断
xiapin
一种负载过渡金属Fe的MOF材料:Fe-NU-1000(见下图),现想对这个团簇进行结构优化,所带电荷和自旋多重度不会判断,想请教各位老师。分子式是C56H54O32FeZr6,用这种材料催化环己烷脱氢。按照文献给出的计算方法和基组分别为:M06-L泛函;CH...
量子化学
250
5
求助:gauss 单点能 中断后还能继续算么?
aaq2800
CCSD(T)单点能计算,算了十几天中断了,如果从头再算,太浪费时间。该输入文件该怎么修改?搜了一下,是不是这样的?请各位大佬给指点一下%rwf=test.rwf%chk=test.chk%mem=50GB%nproc=24#CCSD(T)/def2tzvpg...
量子化学
450
9
荧光或者磷光发射过程,存在MLCT、LLCT、LMCT、LC、MC等过程吗?
simiamest
我查里一些帖子,电子被激发的过程会出现题目里说的那几种情况,那电子从激发态退激的过程,比如说荧光或者磷光的发光过程,也讲MLCT、LLCT、LMCT、LC、MC这些概念吗?
量子化学
300
6
关于mac版本的高斯
lastzealot
官网上查到有mac版本的高斯,请问谁在mac上用高斯做计算呢?能否交流一下心得?如果答疑解惑,可重谢哦~~
量子化学
250
5
QST2优化三天了,还没有结束,帮忙看下有没有问题
sealanlan
优化一个分子闭环到开环的过渡态,第一次选择使用qst2进行优化,三天了,还没有跑完,我是应该截取中间态来做opt=ts呢?还是继续让它跑着呢?输出log文件见附件。
量子化学
150
3
两个结构类似的分子做柔性势能面扫描,结果差别很大,可否帮忙看下怎么回事?
sealanlan
两个类似的分子,固定开环两个碳原子之间的距离,用关键词opt=modredundant,做开环反应的柔性势能面扫描,结果差别很大,可否帮忙看下怎么回事?第一个分子能够从闭...
量子化学
250
5
求助spartan14及以上版本-软件 crack
org-chemist
求助spartan14及以上版本-软件crack!!!
量子化学
200
4
求助一个带我的大神做XTB
poly花
求助一个大神带我做XTB,Linux系统下跑,已经装了虚拟机了,但是还是一脸蒙
量子化学
200
4
反应优先级的确定
清远常山
本人以前是做无机材料,现在转做有机,请教各位大神一个问题,下图中的有机物,H将会被S、O2和O-攻击,但是首先会和谁发生反应呢?是否可以通过Multiwfn计算进行预判呢?具体怎么做呢?氮也有类似的问题,O2和O-谁会先攻击氮呢?请各位大神指点迷津
量子化学
150
3
使用TS获得过渡态,虚频数值与文献能对上,但是虚频振动方面相反,求助!
judd_trump
本人新手,对照文献学习,使用TS获得过渡态,虚频数值与文献能对上,但是虚频振动方面相反,求助!感谢大佬帮助
量子化学
200
4
求一份windows 10操作系统的gaussview
lymyan
求助windows10操作系统的gaussview,哪位大佬分享一下,谢谢?
量子化学
250
5
请问Gaussian能模拟高温下芳香烃的振动分辨发射光谱吗?
YJ天下大同
如标题,刚接触量子化学,请问Gaussian能模拟高温下(1000K)芳香烃的振动分辨发射光谱(荧光光谱)吗?我看到的文献说得很模糊,或者书记0k下的光谱结果。如果可以模拟,高温效应是在哪里设置的呢?
量子化学
400
8
Gaussian计算出现多个freq是负数,虚频怎么办
afko9138
Gaussian计算出现多个freq是负数,虚频怎么办
量子化学
150
3
高斯计算如何获得电化学反应吉布斯自由能
江汉秋影燕
求教一个高斯计算的问题,请各位大佬帮帮忙,谢谢文章中写的吉布斯在自由能是由:G=Eelect+EZPE+PV+U298−TS298;其中Eelect和EZPE分别是电子总能量和零点能量。PV代表压强−体积积,后两项是有限的温度对内能和熵...
量子化学
300
6
Gaussian可以用于计算四氧化三铁的电子能级轨道吗?
大帝拉乌斯
Gaussian可以用于计算四氧化三铁的电子能级轨道吗?如果可以基组和DFT方法应该如何选择?
量子化学
250
5
文件类型转换求助
单单单1234
请问如何计算Mayer键序,如何将wfn格式转换为fch文件?
量子化学
250
5
量子化学计算券
知止而后动
手里一张测试狗的代金券,感兴趣的私聊哦!
量子化学
2700
54
关于Mayer键序计算问题
单单单1234
计算Mayer键序,基组不能带弥散函数。如果去掉弥散函数重新计算,我试了很多还是没有成功。请问高斯的输入文件关键词需要用那些算的更快一点呢?
量子化学
150
3
超算中心的账号可以自己安装高斯吗
zxy7909
如题,请教大家如何装
量子化学
250
5
cc-pwCVTZ-PP基组
gscn
最近在文章中能够看到Zr使用的是cc-pwCVTZ-PP基组,请问这种基组gaussian怎么设置?
量子化学
300
6
请教一下比较分子能量时候考虑负号吗?
iamlongwei
各位高人:小弟不才,一直在摸索高斯gaussian的计算,在能量输出部分出现了疑惑,输出文档的末端HF是电子的总能量,内能是电子能量+内能校正项,一般是负数的。我在一本教科书上反复看到,负数小的反而能量高?这是对的吗?比如:经过计算得到A分子的稳定构象内能是-...
量子化学
150
3
HOMO-LUMO gap是负值
1240906hs
想请教大神,用高斯计算后得到的体系HOMO能量是-2.97ev,LUMO能量是-4.11ev,gap是-1.14ev。为什么gap是负值呢?这表明什么呢?谢谢大神!
量子化学
300
6
Gaussian 16输出文件转换
白日做梦3
请问各位大神如何将Gaussian16输出文件(.log或.out)转换输入文件(.gif)?我想查看是具体使用了什么样泛函以及输入文件的具体内容。
量子化学
150
3
求助---高斯计算锂离子的单点能
lp00
小白用高斯计算Li离子的单点能量。总是在一开始就报错,请高手指导提交计算的文件%nprocshared=24%mem=4GB%chk=C:\\Users\\Administrator\\Desktop\\LiDFAMB-bindingenergy\\energ...
量子化学
300
6
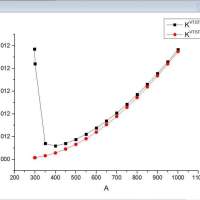
反应速率常数变化异常是为什么呢?
单单单1234
请问反应速率常数随温度变化图,在低温的时候变化异常可能是什么原因呢?谢谢[Lasteditedby单单单1234on2021-3-2at17:15]
量子化学
150
3
能否帮忙排下丙三醇、山梨醇、葡萄糖、蔗糖、脂肪酸钠的极性大小?
chemlaw
能否帮忙排下在水溶液中的丙三醇、山梨醇、葡萄糖、蔗糖、脂肪酸钠的极性大小?可以用查出来的偶极矩数据来排极性大小,也可以通过量子化学计算的方式来排。注:水溶液中,葡萄糖几乎都以环式存在,链式极少。
量子化学
150
3
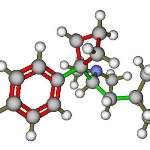
N2和NO电子跃迁矩求助
梁子XDU
本人量化小白一个,也没有太多时间学习量化理论。因为方向不偏向这些。只是需要用到一些基础数据,如电子跃迁矩。目前知道Molpro软件能计算双原子分子的电子跃迁矩,现需要下图...
量子化学
2000
40
活动开始,高性能计算机时资源
李先生阿
北京云计算中心,高性能资源活动开始。扫码填写申请免费核时资源的账号,手慢一些怕是要看不到贴了。图片发不了超清,不好意思哦各位。
量子化学
250
5
vmd使用教程,求,急
Liumoshu
vmd使用教程,求,急
量子化学
150
3
13463
‹‹
1
2
3
4
5
6
››
... 449
板块导航
网络生活
育儿交流
健康生活
有奖问答
资源共享
课件资源
试题资源
化学化工
有机
高分子
无机物化
分析
催化
工艺技术
化工设备
化工
精细化工
电化学
环境
专业学科
机械
物理
数学
农林
食品
地学
能源
信息科学
理工农林
科研生活
博后之家
专业外语
外语学习
导师招生
找工作
招聘信息
考研
考博
公务员
生物医药
新药研发
药学
药品生产
分子生物
微生物
动植物
生物科学
医学
材料
材料
材料工程
微米纳米
晶体
金属
非金属
生物材料
功能材料
复合材料
计算模拟
第一原理
量子化学
计算模拟
分子模拟
仿真模拟
程序语言
学术交流
论文投稿
基金申请
学术会议
出国留学
留学生活
公派出国
访问学者
海外博后
留学DIY
签证指南
出国考试
海外院所
注册执考
化工工程师
执业药师
执业医师
环境工程师
会计师
注册考试
24小时热帖
换一批
小孩7岁,上一年级,不自信,学习较差
19
估计今年青基又没戏
3
杭州国企和浙江高校如何选择?
19
博后换方向可行吗?
3
家乡二本高校/沿海传统私企,如何抉择?
12
应助之星
nono2009
+关注
czyzsu
+关注
topedit
+关注
591950582
+关注
sleethail2
+关注
下载小木虫APP
与700万科研达人随时交流
二维码
IOS
安卓
欢迎监督和反馈
:小木虫仅提供交流平台,不对该内容负责。
欢迎协助我们监督管理,共同维护互联网健康,违规贴举报删除请联系邮箱:libolin3@tal.com 或者 QQ:64901448
(点此查看侵权举报方式)
我们保证在7个工作日内给予处理和答复,谢谢您的监督。
©2001-2024 muchong.com,小木虫
京ICP备16008351号
京公网安备 11010802022153号
Copyright © 2001-2024 muchong.com, All Rights Reserved. 小木虫 版权所有