ALN的能带. 发现EF 不等于0
jackwangee
Hi,大家好我正在测试用QE做ALN的bandstructure.发现EF不等于0!但是大部分文献里是EF=0.没办法贴图。贴输入文件。&CONTROLcalculation="nscf"max_seconds=8.64000e+0...
第一原理
- 200
- 4
ALN的能带. 发现EF 不等于0
jackwangee
Hi,大家好我正在测试用QE做ALN的bandstructure.发现EF不等于0!但是大部分文献里是EF=0.没办法贴图。贴输入文件。&CONTROLcalculation="nscf"max_seconds=8.64000e+0...
第一原理
- 200
- 4
请一下各位老师:
lastnumber
我在计算cohp时,出现正交化警告,导致一直都把chargespilling降不下去,cohp手册里基本参数都试过了。不知道如何解决,有经验的就告诉我一下该如何处理。先在此谢谢各位了
第一原理
- 1950
- 39

澳门大学 材料模拟与计算 科研助理招聘 (年薪15万澳门币)
dielectric1
澳门大学蔡永青课题组招聘DFT计算研究助理1名(硕士毕业即可,年薪15万澳门币),要求申请者至少在下列领域之一有相关研究经验:1)声子;2)含时密度泛函以及激发态;3)高通量计算以及人工智能;4)相场模拟;5)铁电压电;6)热电/热导。要求熟练掌握一门第一性原...
第一原理
- 350
- 7

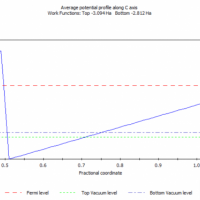

Vasp计算Pt-111电催化HER的吸附H吉普斯自由能变化
sunnyrainy
计算新手一枚,最近用Vasp计算析氢反应HER的吸附H吉普斯自由能变化△G。使用GGAPBEDFT+D3,模拟计算Pt-111对H的吸附,4X4X4slab,固定底部两层,使用公式:△G=E(H吸附)+0.24eV。调整了很多输入文件参数,但是,我计算的△G均...
第一原理
- 400
- 8