

gaussian计算有机物NICS值判断有机物芳香性,有一些问题向各位大佬咨询一下。
tyj94335625
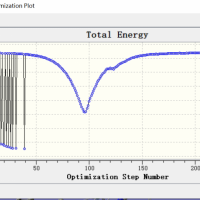
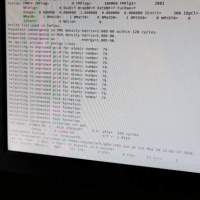
是这样的。我照着网上查找的一些资料用gaussian09计算我使用的有机物的NICS值。关键词输入NMR=GIAO,使用的基组是B3LYP/6-31+dp。考虑到我使用的有机物分子量很大,一个分子有好几个环,所以我不知道怎么确定有机物的平面,就只好先计算NIC...
量子化学
- 200
- 4







计算超精细耦合常数
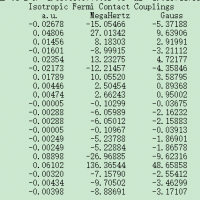
1259483770
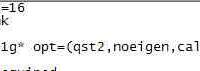
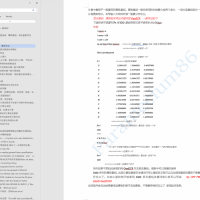
请问各位大佬,我通过Gaussian计算出来了IsotropicFermiContactCouplings,我要如何通过它来确定超精细耦合常数。请各位指教!截图.png
量子化学
- 200
- 4
计算超精细耦合常数
1259483770
请问各位大佬,我通过Gaussian计算出来了IsotropicFermiContactCouplings,我要如何通过它来确定超精细耦合常数。请各位指教!截图.png
量子化学
- 400
- 8






